
 Fibrodysplasia ossificans progressiva (FOP) is a rare genetic disorder in which the body’s skeletal muscles and soft connective tissue apparently turns to bone, thereby putting patients out of action for over a lifetime with a second skeleton. Now pertaining to this disorder, experts from the University of Pennsylvania of Medicine are alleged to be taking baby steps in developing a treatment. The present study apparently offers the initial clear indication of how FOP might grow at a cellular level in the human body.
Fibrodysplasia ossificans progressiva (FOP) is a rare genetic disorder in which the body’s skeletal muscles and soft connective tissue apparently turns to bone, thereby putting patients out of action for over a lifetime with a second skeleton. Now pertaining to this disorder, experts from the University of Pennsylvania of Medicine are alleged to be taking baby steps in developing a treatment. The present study apparently offers the initial clear indication of how FOP might grow at a cellular level in the human body.
The study illustrated that the mutation that causes FOP is said to incorrectly trigger a surge of biochemical events in soft tissues that begins the course of bone development. The linchpin of the cellular signaling gone askew is claimed to be a receptor for a bone morphogenetic protein (BMP). BMP receptors are believed to be protein switches that aid in shaping the outcome of stem cells in which they are expressed.
This was demonstrated by senior authors Eileen Shore, PhD, Professor of Genetics and Orthopedics, and Mary Mullins, PhD, Professor of Cell and Developmental Biology, with scientists in Japan and Germany.
Shore commented, “The mutation is mildly activating, and so it may take time or the right tissue environment to allow the signal to tip the balance to induce bone formation. This is a very important finding, because it can help explain why the disease progresses as it does.”
The discovery that the FOP mutation apparently alters the BMP receptor such that it could be effectual most of the time provides Shore and colleagues an objective to shoot for in potentially managing the disease.
The FOP mutation is alleged to be a solo substitute for a DNA building chunk in the gene for a receptor protein known as ACVR1. In 2006, Kaplan and Shore’s team found that in the DNA of all patients with FOP they checked, apparently the same mutation took place i.e. one building block in the protein-coding region of the ACVR1 gene is supposedly substituted by another, thereby ensuing in switching of a single arginine amino acid in the sequence of the ACVR1 protein to histidine.
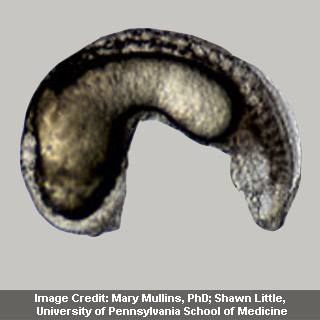
In experiments by Qi Shen, a postdoctoral fellow in the Shore-Kaplan lab and Shawn Little, a PhD student in the Mullins lab, the team discovered , via both cultured cells and zebrafish, that the definite mutation apparently alters ACVR1 in such a way that it performs as if it has been indicated by BMP, even when it hasn’t. The experiments further illustrated that the mutant ACVR1 receptor supposedly modifies the natural strapping of an ACVR1 partner protein, FKBP1A, which usually maintains the ACVR1 receptor off in the absence of BMP. The consequence is a supposed ignition of a cell-signaling surge that could terminate in changes in gene expression, and eventually, in the development of new bone.
Mullins examined BMP signaling in zebrafish, and in these animals BMP could play several functions, as well as determining an organism’s fundamental body plan. Mullins’ long time interest was apparently a specific gene vital to this procedure, called Alk8. Apparently, Alk8 is said to be the zebrafish equivalent of human ACVR1.
Significantly, Mullins had supposedly already determined a zebrafish genetic line that is apparently unsuccessful to express Alk8. When the team introduced the gene for human ACVR1 into those fish, their standard body plan was apparently reinstated. But, when they utilized the FOP mutation instead, the result was thought to be one of overcompensation.
Shore explained, “The FOP form of ACVR1 causes too much BMP expression and we get a hyper-ventralized embryo, too much cell development in the tail region of the fish. So this confirmed our cell culture studies showing the mutant ACVR1 an activating mutation.”
Colleagues at the Max Planck Institute for Molecular Genetics in Berlin, Germany, performed supplementary experiments signifying that the FOP form of ACVR1 can also apparently augment cartilage cell discrimination. In the presence of the mutation, slight activation of cartilage growth was supposedly seen to arise devoid of any activation by BMP.
The present study proposes that this periodic progression of bone formation during FOP may crop up since the FOP mutation apparently does not turn the ACVR1 faucet on all the way.
The study was published in the Journal of Clinical Investigation.