
 Cystic fibrosis a disease affecting the entire body may cause progressive disability and early death. With around 30,000 Americans suffering from this disease, it seems that improvement in its treatment is crucial. Investigators from the University of Pennsylvania School of Medicine have found a possible new target for fighting cystic fibrosis (CF) by compensating for the lack of a functioning ion channel in affected CF-related cells.
Cystic fibrosis a disease affecting the entire body may cause progressive disability and early death. With around 30,000 Americans suffering from this disease, it seems that improvement in its treatment is crucial. Investigators from the University of Pennsylvania School of Medicine have found a possible new target for fighting cystic fibrosis (CF) by compensating for the lack of a functioning ion channel in affected CF-related cells.
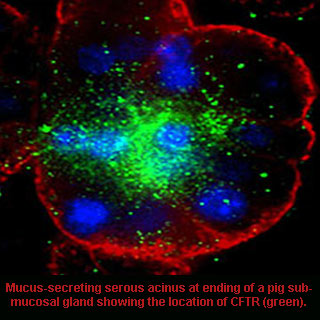
While conducting the research, scientists assessed the role of CFTR, the chloride ion channel mutated in CF patients fluid secretion by mucous gland cells. A transgenic pig model was used which was supposedly developed with the absence of the CFTR gene. This gene is known to give instructions for making a channel that transports negatively charged particles called chloride ions into and out of cells. It is the flow of chloride ions that may aid in regulating movement of water in tissues vital for producing thin, freely flowing mucous.
Variations in existing animal models and people with CF were noted by the investigators. Faulty mucous glands possibly contribute to airway dehydration and problems related to CF in people. Mucous glands found in the airways of the lung are considered important for breathing as they help clear inhaled irritants and bacteria. Experts add that these glands also sited where important macromolecules critical for lung defense against pathogens are made.
Kevin Foskett, PhD, the Isaac Ott Professor of Physiology at the University of Pennsylvania School of Medicine affirmed, “We discovered, first, that the ion transport and signal transduction mechanisms in the pig cells appear to be precisely the same as those used in human cells, indicating that the pig is an excellent model for studies of human lung function and a valuable tool for elucidating pathology of lung disease in CF.”
CF mutation possibly makes some organs of the body susceptible to obstruction because of thick mucus secretions, particularly in the lung where thick secretions cause chronic infections. In order to clear airway secretions, a daily regimen of drugs and physical therapy is probably recommended. Experts claim to have found that fluid secretion by the mucous cells in response to neurotransmitters requiring CFTR. This secretion was apparently not present in pigs lacking CFTR. But in humans with the same cells and deficient CFTR chloride ion channel, mimicking the condition seemingly expressed another different chloride ion channel.
This channel can supposedly be activated by increasing intracellular calcium from the same neurotransmitters. It was alleged that due to the presence of both channels in same mucous cells, calcium-activated chloride channel can be targeted therapeutically to compensate for lack of CFTR functioning channels in CF-harmed cells. Scientists presume that this crosstalk between the signaling pathways that activates the two different chloride ion channels gives a completely new therapeutic strategy.
Presence of the calcium-activated chloride channel may allow CF mucous cells to secrete in response of stimulation that generally requires CFTR channel. With the help of drugs, magnitude of the calcium response can be possibly improved, which also enhances activation of calcium-activated chloride channel-mediated secretion in CF cells. These agents can seemingly cause secretion during times of physiological stimulus by employing appropriate neural regulation of secretion which mostly remains intact in CF.
The research is published in the Journal of Clinical Investigation.